idopNetwork_vignette
Ang Dong
06/07/2023
Source:vignettes/idopNetwork_vignette.Rmd
idopNetwork_vignette.Rmd
library(idopNetwork)
backup_options <- options()
#load pre-computered results
test_result = idopNetwork:::test_resultAbout
idopNetwork is packed as a cartographic tool that performs power curve fitting, classification, variable selection, microbial abundance decomposition, and network visualization based on microbial 16S rRNA gene sequencing metadata.
For complete details on the use and execution of this protocol, please refer to Chen et al and Cao et al.
1. Input data
Before running idopNetwork, user need to provide datasets, and they should be cleaned and merged to exactly the same format of the example data.
1.1 gut microbe OTUs data
Microbe Operational taxonomic unit dataset must have first column as IDs for microbes.
| name | GQ_UC1 | GQ_UC3 | GQ_UC4 | GQ_UC5 |
|---|---|---|---|---|
| Escherichia_coli | 176 | 1701 | 28506 | 177 |
| Arthrobacter_oxydans | 3 | 215 | 1578 | 1 |
| Ruminococcus_gnavus | 744 | 0 | 4 | 733 |
| Bacteroides_plebeius | 271 | 4401 | 7 | 8394 |
| Bacteroides_pyogenes | 0 | 15 | 0 | 0 |
| Clostridium_tertium | 305 | 1 | 1 | 270 |
| Bacteroides_stercoris | 24 | 0 | 0 | 11 |
| Flavisolibacter_ginsengisoli | 0 | 123 | 831 | 0 |
| Bacteroides_massiliensis | 0 | 6098 | 6 | 0 |
| Prevotella_heparinolytica | 0 | 6 | 0 | 0 |
1.2 mustard microbe OTUs data
| ID | 8_0149.leaf | 8_0157.leaf | 8_0158.leaf | 8_0160.leaf | |
|---|---|---|---|---|---|
| 2 | OTU_1 | 3 | 2 | 59 | 268 |
| 3 | OTU_2 | 8 | 13 | 1890 | 44 |
| 4 | OTU_3 | 5485 | 3302 | 4522 | 6581 |
| 5 | OTU_4 | 42 | 351 | 7 | 4 |
| 6 | OTU_5 | 16 | 2002 | 24 | 43 |
| 7 | OTU_6 | 1645 | 1170 | 10636 | 31686 |
| 8 | OTU_7 | 4050 | 4094 | 13 | 66 |
| 9 | OTU_8 | 6 | 282 | 124 | 27 |
| 10 | OTU_9 | 102 | 5 | 10 | 18 |
| 11 | OTU_10 | 25 | 1393 | 35 | 19 |
| JAM_E4.leaf | 8_0149.root | 8_0157.root | 8_0158.root | 8_0160.root | |
|---|---|---|---|---|---|
| 2 | 7 | 28861 | 122 | 1 | 9 |
| 3 | 4 | 11618 | 19 | 14 | 1894 |
| 4 | 4981 | 481 | 755 | 17914 | 15998 |
| 5 | 622 | 862 | 1072 | 11 | 103 |
| 6 | 10014 | 447 | 758 | 19 | 289 |
| 7 | 577 | 1010 | 490 | 25030 | 29443 |
| 8 | 2233 | 13 | 5575 | 26 | 64 |
| 9 | 19 | 3348 | 36 | 33 | 46 |
| 10 | 362 | 6269 | 228 | 2 | 459 |
| 11 | 3071 | 4 | 6454 | 6 | 234 |
2. power curve fitting
The first major step in our idopNetwork reconstruction is to fit allometric growth curves to the data using the power function. This is easily done by using the related function power_fit. This function needs cleaned dataset as input and will return fitted OTUs for given dataset. Then the fitted output with original dataset can be transfer into function power_equation_plot for quick visualization
df = data_cleaning(gut_microbe)
result1 = power_equation_fit(df)
power_equation_plot(result1)
3. Functional clustering
In this step we implement the power equation into functional clustering to detect different microbial modules. If after clustering there are still too many microbes within a certain module for network reconstruction, we can rerun functional clustering to further classify this module into distinct submodules.
3.1 mean curve
we fit mean vector of each cluster center by power equation(assume k=3)
matplot(t(power_equation(x = 1:30, matrix(c(2,1,3,0.2,0.5,-0.5),nrow = 3, ncol = 2))),
type = "l",
xlab = "time",
ylab = "population")
legend("topright",
c("cluster 1", "cluster 2", "cluster 3"),
lty = c(1,2,3),
col = c(1,2,3),
box.lwd = 0)
3.2 covariance matrix
we fit covariance matrix of multivariate normal distribution with SAD1, it can be showed as
get_SAD1_covmatrix(c(2,0.5), n = 5)
#> [,1] [,2] [,3] [,4] [,5]
#> [1,] 0.25 0.50 1.00 2.00 4.00
#> [2,] 0.50 1.25 2.50 5.00 10.00
#> [3,] 1.00 2.50 5.25 10.50 21.00
#> [4,] 2.00 5.00 10.50 21.25 42.50
#> [5,] 4.00 10.00 21.00 42.50 85.253.3 initial parameters
we can check initial parameters (k=4)
get_par_int(X = log10(df+1), k = 4, times = as.numeric(log10(colSums(df)+1)))
#> Error in nls(y ~ a * x^b, start = list(a = a, b = b), control = nls.control(maxiter = 1000, :
#> number of iterations exceeded maximum of 1000
#> Error in nls(y ~ a * x^b, start = list(a = a, b = b), control = nls.control(maxiter = 1000, :
#> number of iterations exceeded maximum of 1000
#> Error in nls(y ~ a * x^b, start = list(a = a, b = b), control = nls.control(maxiter = 1000, :
#> number of iterations exceeded maximum of 1000
#> Error in nls(y ~ a * x^b, start = list(a = a, b = b), control = nls.control(maxiter = 1000, :
#> number of iterations exceeded maximum of 1000
#> Error in nls(y ~ a * x^b, start = list(a = a, b = b), control = nls.control(maxiter = 1000, :
#> number of iterations exceeded maximum of 1000
#> Error in nls(y ~ a * x^b, start = list(a = a, b = b), control = nls.control(maxiter = 1000, :
#> number of iterations exceeded maximum of 1000
#> Error in nls(y ~ a * x^b, start = list(a = a, b = b), control = nls.control(maxiter = 1000, :
#> number of iterations exceeded maximum of 1000
#> $initial_cov_params
#> [1] 0.50000 1.09618
#>
#> $initial_mu_params
#> a b
#> [1,] 1.222931e+15 -21.7299294
#> [2,] 2.241187e-35 50.4618037
#> [3,] 1.275766e+01 -0.8462029
#> [4,] 1.930699e+02 -3.6047139
#>
#> $initial_probibality
#>
#> 1 2 3 4
#> 0.24590164 0.27868852 0.06557377 0.40983607
#use kmeans to get initial centers
tmp = kmeans(log10(df+1),4)$centers
tmp2 = power_equation_fit(tmp, trans = NULL)
power_equation_plot(tmp2, label = NULL,n = 4)
3.4 functional clustering (power-equation,SAD1)
idopNetowrk already wrapped the mean curve modelling, covariance
matrix modelling and likelihood ratio calculation into a function
fun_clu().
options(max.print = 10)
fun_clu(result1$original_data, k = 3, iter.max = 5)
#> Error in nls(y ~ a * x^b, start = list(a = a, b = b), control = nls.control(maxiter = 1000, :
#> number of iterations exceeded maximum of 1000
#> initial value 1224.701250
#> final value 1224.701250
#> converged
#>
#> iter = 1
#> Log-Likelihood = 1229.444
#> initial value 1228.240624
#> final value 1228.240624
#> converged
#>
#> iter = 2
#> Log-Likelihood = 1225.461
#> initial value 1224.705979
#> final value 1224.705979
#> converged
#>
#> iter = 3
#> Log-Likelihood = 1226.112
#> initial value 1225.588016
#> final value 1225.588016
#> converged
#>
#> iter = 4
#> Log-Likelihood = 1224.793
#> initial value 1224.655751
#> final value 1224.655751
#> converged
#>
#> iter = 5
#> Log-Likelihood = 1224.97
#> initial value 1224.822485
#> final value 1224.822485
#> converged
#>
#> iter = 6
#> Log-Likelihood = 1227.894
#> $cluster_number
#> [1] 3
#>
#> $Log_likelihodd
#> [1] 1227.894
#>
#> $AIC
#> [1] 2475.787
#>
#> $BIC
#> [1] 2494.499
#>
#> $cov_par
#> [1] 0.500000 1.098059
#>
#> $mu_par
#> [,1] [,2]
#> [1,] 2.161219e-15 21.401464
#> [2,] 4.804131e+01 -1.686739
#> [3,] 2.210693e+16 -23.629600
#>
#> $probibality
#> [1] 0.57603917 0.07110126 0.35285957
#>
#> $omega
#> [,1] [,2] [,3]
#> [1,] 2.536304e-01 3.140715e-04 7.460555e-01
#> [2,] 4.400950e-01 2.237826e-04 5.596812e-01
#> [3,] 5.594828e-01 1.463402e-06 4.405157e-01
#> [ reached getOption("max.print") -- omitted 45 rows ]
#>
#> $cluster
#> MC_UC3 MC_UC4 YZJC_UC3 GQ_UC3 ZC_UC3 MC_UC2 GQ_UC1 GQ_UC4 HJC_UC3 ZC_UC4
#> HJC_UC1 HJC_UC4 HC_UC2 ZC_UC1 HC_UC4 JJC_UC2 HJC_UC2 MC_UC1 GQ_UC5 ZC_UC5
#> apply(omega, 1, which.max)
#> [ reached 'max' / getOption("max.print") -- omitted 48 rows ]
#>
#> $cluster2
#> MC_UC3 MC_UC4 YZJC_UC3 GQ_UC3 ZC_UC3 MC_UC2 GQ_UC1 GQ_UC4 HJC_UC3 ZC_UC4
#> HJC_UC1 HJC_UC4 HC_UC2 ZC_UC1 HC_UC4 JJC_UC2 HJC_UC2 MC_UC1 GQ_UC5 ZC_UC5
#> apply(omega, 1, which.max)
#> [ reached 'max' / getOption("max.print") -- omitted 48 rows ]
#>
#> [ reached getOption("max.print") -- omitted 2 entries ]Usually we use multithread to calcuation k = 2-n and then to decide
best k , fun_clu_BIC uses BIC to select best cluster number
by default.
result2 = fun_clu_parallel(result1$original_data, start = 2, end = 5)
best.k = which.min(sapply( result2 , "[[" , 'BIC' )) + 1 #skipped k = 1
best.k
#> [1] 7
fun_clu_BIC(result = result2)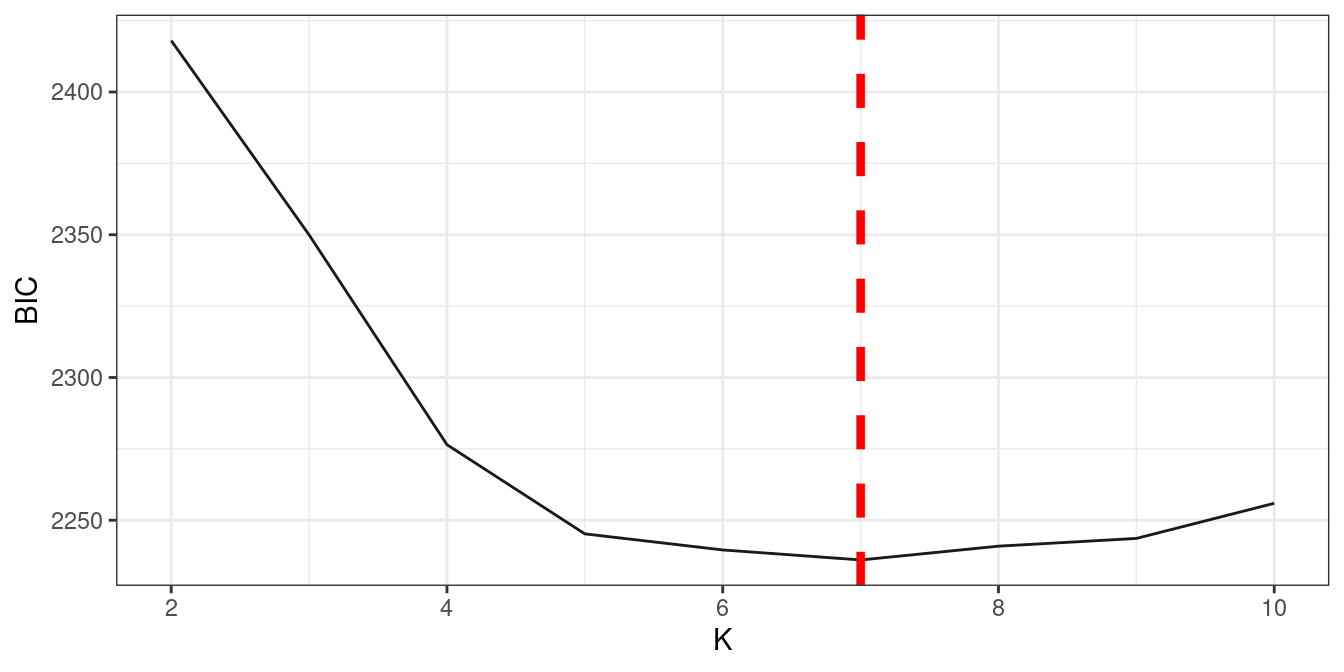
#we can direct give other k value
fun_clu_plot(result = result2, best.k = best.k)
3.5 bi-functional clustering (power-equation,biSAD1)
In this part we select part of mustard data for demonstration purpose.
data("mustard_microbe")
df2 = data_cleaning(mustard_microbe, x = 160)res_l = power_equation_fit(df2[,1:5]
res_r = power_equation_fit(df2[,89:95])
res1 = data_match(result1 = res_l, result2 = res_r)
res2 = bifun_clu_parallel(data1 = res1$dataset1$original_data,
data2 = res1$dataset2$original_data,
Time1 = res1$dataset1$Time,
Time2 = res1$dataset2$Time,
start = 2,
end = 10,
thread = 9,
iter.max = 10)
res2 = bifun_clu_parallel(data1 = res1$dataset1$original_data,
data2 = res1$dataset2$original_data,
Time1 = res1$dataset1$Time,
Time2 = res1$dataset2$Time,
start = 2,
end = 10,
thread = 9,
iter.max = 10)
fun_clu_BIC(result = res2)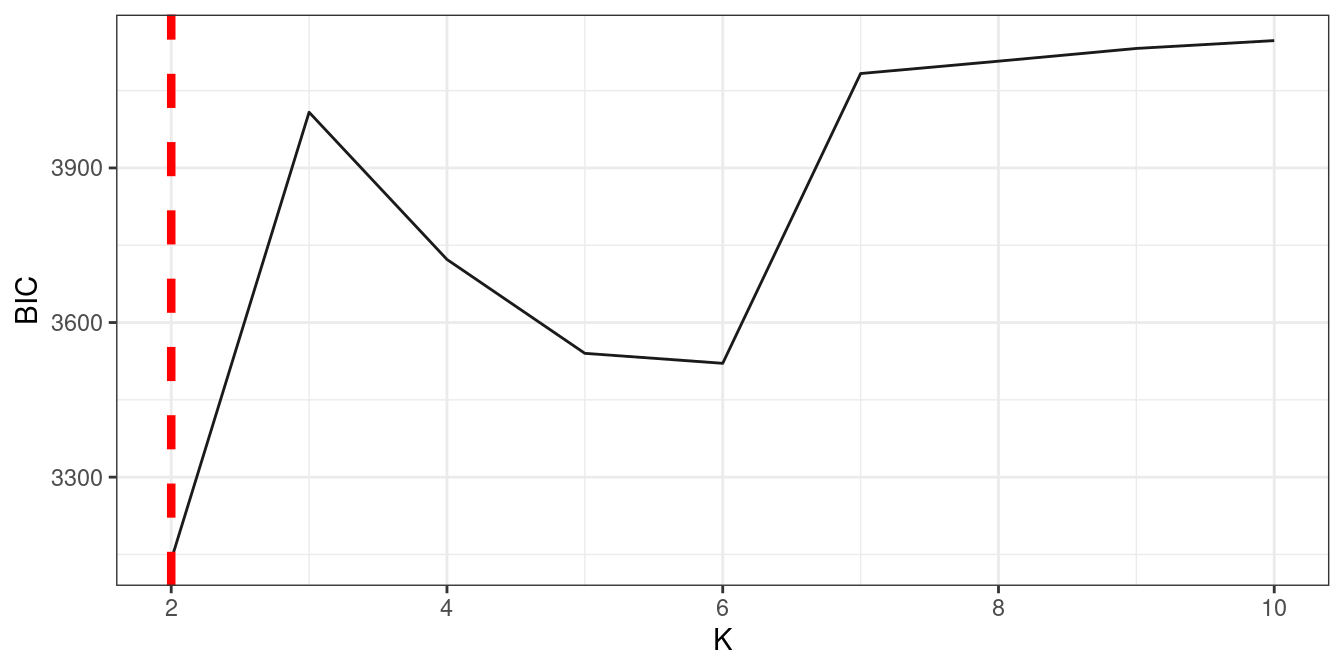
#we can set best.k directly
bifun_clu_plot(result = res2, best.k = 3, color1 = "#C060A1", color2 = "#59C1BD")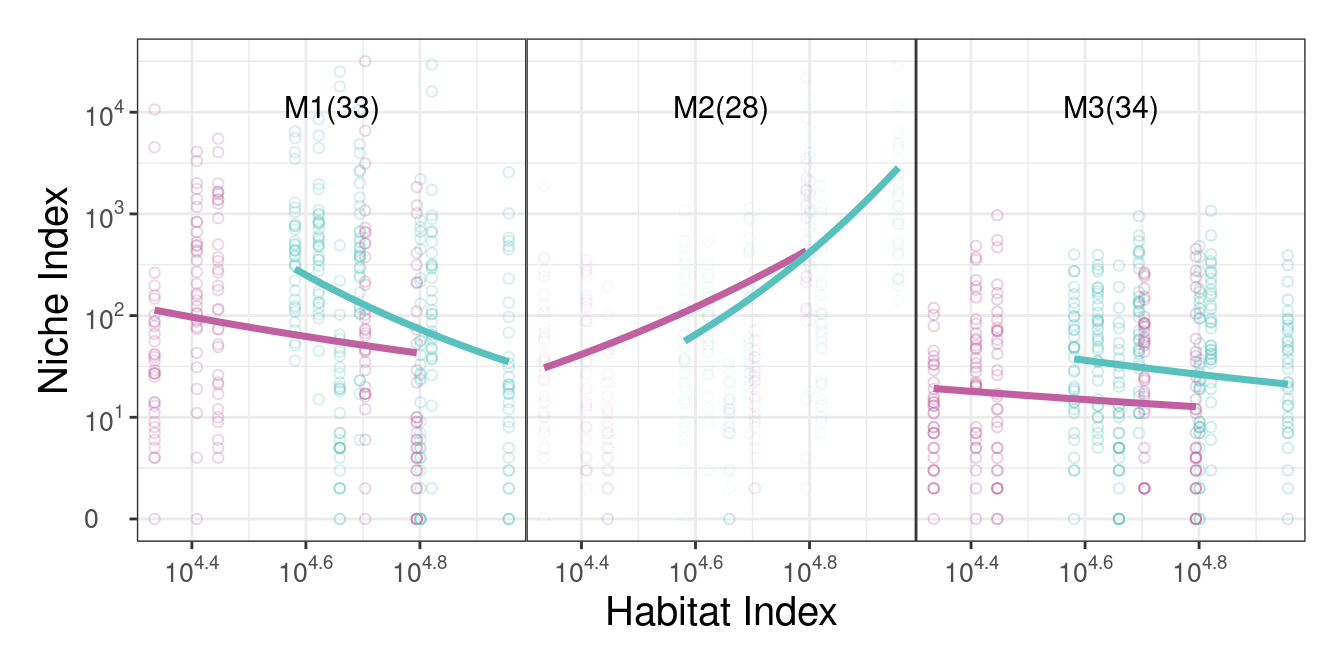
3.6 sub-clustering
Sometimes a module is still too large for network reconstruction, which is determined by Dunbar’s number, we can further cluster it into sub-modules.
res3 = bifun_clu_convert(res2, best.k = 3)
large.module = order(sapply(res3$a$Module.all,nrow))[5]
res_suba = fun_clu_select(result_fit = res1$dataset1, result_funclu = res3$a, i = large.module)
res_subb = fun_clu_select(result_fit = res1$dataset2, result_funclu = res3$b, i = large.module)
dfsuba_l = power_equation_fit(res_suba$original_data)
dfsubb_r = power_equation_fit(res_subb$original_data)
ressub1 = data_match(result1 = dfsuba_l, result2 = dfsubb_r)
ressub2 = bifun_clu_parallel(data1 = ressub1$dataset1$original_data,
data2 = ressub1$dataset2$original_data,
Time1 = ressub1$dataset1$Time,
Time2 = ressub1$dataset2$Time,
start = 2,
end = 5,
iter.max = 3)
ressub2 = test_result$d2_subcluster
fun_clu_BIC(result = ressub2)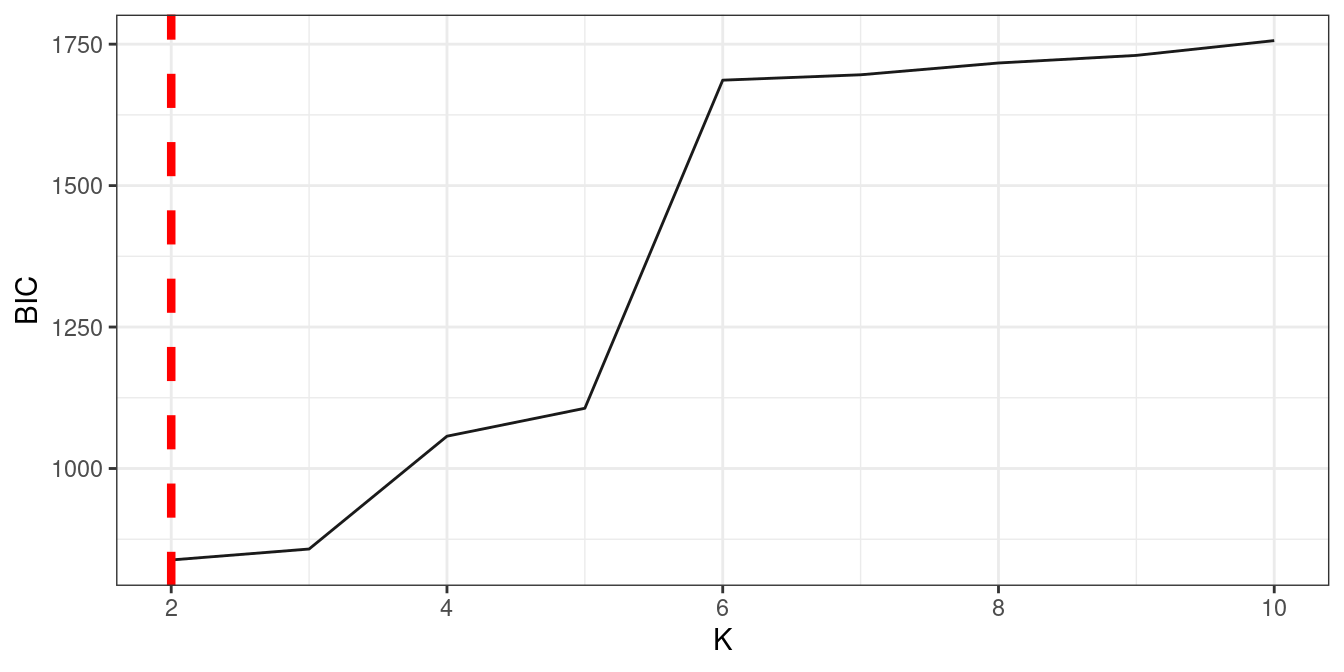
bifun_clu_plot(result = ressub2, best.k = 2, color1 = "#C060A1", color2 = "#59C1BD", degree = 1)
4. LASSO-based variable selection
idopNetwork implements a LASSO-based procedure to choose a small set
of the most significant microbes/module that links with a given
microbes/modules. get_interaction()return a compound list
contain the target microbe/module name, the most relevant
Modules/microbes names and the coefficients.
4.1 For Modules
result3 = fun_clu_convert(result2,best.k = best.k)
df.module = result3$original_data
get_interaction(df.module,1)
#> $ind.name
#> [1] "M1"
#>
#> $dep.name
#> [1] "M5"
#>
#> $coefficient
#> [1] 5.9640784.2 For Microbes
#we can the microbial relationship in Module1
df.M1 = result3$Module.all$`1`
get_interaction(df.M1,1)
#> Warning: Option grouped=FALSE enforced in cv.glmnet, since < 3 observations per
#> fold
#> Warning: Option grouped=FALSE enforced in cv.glmnet, since < 3 observations per
#> fold
#> Warning: Option grouped=FALSE enforced in cv.glmnet, since < 3 observations per
#> fold
#> $ind.name
#> [1] "Arthrobacter_oxydans"
#>
#> $dep.name
#> [1] "Fusobacterium_necrophorum"
#>
#> $coefficient
#> [1] 0.082635255 qdODE solving
qdODE system is build based on variable selection results, it has unique ability to decompose the observed module/microbe abundance level into its independent component and dependent component, which can be used for inferring idopNetwork.
5.1 solving qdODE between modules
options(max.print = 10)
# first we test solving a qdODE
module.relationship = lapply(1:best.k, function(c)get_interaction(df.module,c))
ode.test = qdODE_all(result = result3, relationship = module.relationship, 1, maxit = 100)
#> iter 10 value 0.004336
#> final value 0.000002
#> converged
# we can view the result
qdODE_plot_base(ode.test)
# then we solve all qdODEs
ode.module = qdODE_parallel(result3)
qdODE_plot_all(ode.module)
5.2 solving qdODE within a module
result_m1 = fun_clu_select(result_fit = result1, result_funclu = result3, i = 1)
ode.M1 = qdODE_parallel(result_m1)
qdODE_plot_all(ode.M1)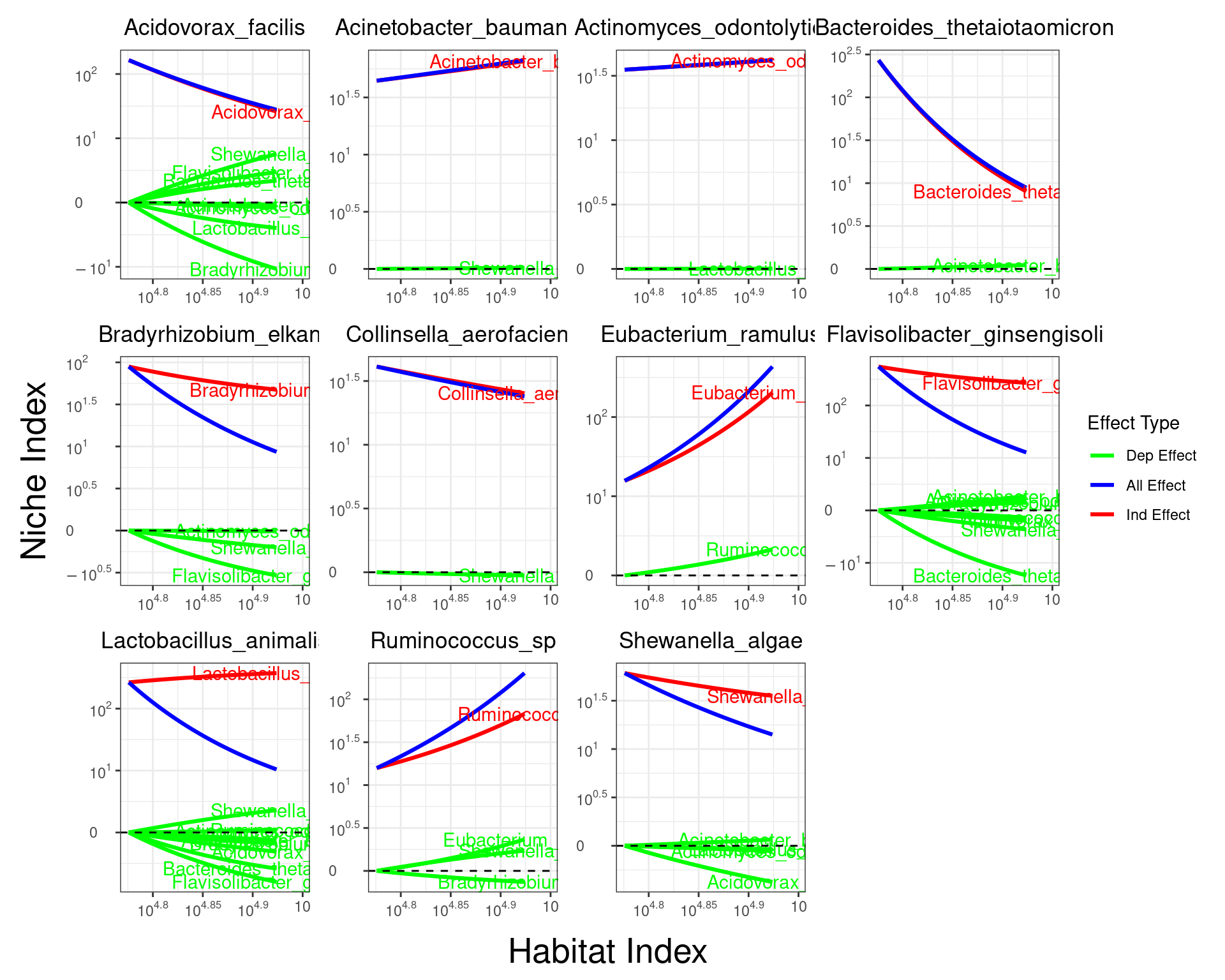
6 idopNetwork reconstruction
The final step of this guide is to visualization the multilayer network, and our package provide network_plot function to easily draw our idopNetwork. We can simply plug previous qdODE results into network_conversion function, and it will convert qdODE result for network visualization
6.1 network between modules
net_module = lapply(ode.module$ode_result, network_conversion)
network_plot(net_module, title = "Module Network")
6.2 network within a module
net_m1 = lapply(ode.M1$ode_result, network_conversion)
network_plot(net_m1, title = "M1 Network")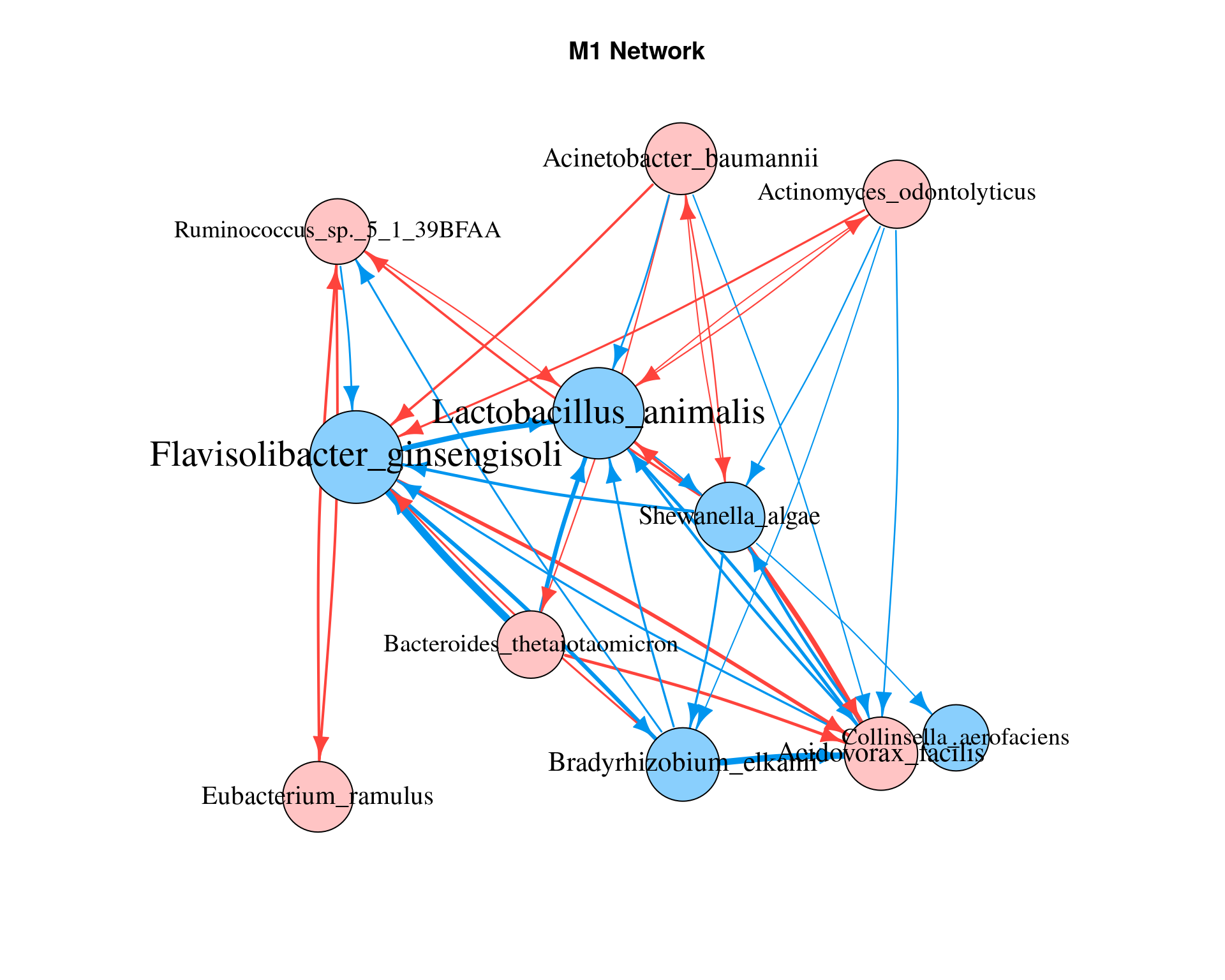
6.3 network comparison
mustard_module_a = qdODE_parallel(res3$a)
mustard_module_b = qdODE_parallel(res3$b)
res_m1a = fun_clu_select(result_fit = res1$dataset1, result_funclu = res3$a, i = 3)
res_m1b = fun_clu_select(result_fit = res1$dataset2, result_funclu = res3$b, i = 3)
mustard_M1a = qdODE_parallel(res_m1a)
mustard_M1b = qdODE_parallel(res_m1b)
mustard_m_a <- lapply(mustard_module_a$ode_result, network_conversion)
mustard_m_b <- lapply(mustard_module_b$ode_result, network_conversion)
#set seed to make same random layout
layout(matrix(c(1,2),1,2,byrow=TRUE))
set.seed(1)
network_plot(mustard_m_a, title = "Module Network a")
set.seed(1)
network_plot(mustard_m_b, title = "Module Network b")
result_m1a = fun_clu_select(result_fit = res1$dataset1, result_funclu = res3$a, i = 1)
result_m1b = fun_clu_select(result_fit = res1$dataset2, result_funclu = res3$b, i = 1)
ode.m1a = qdODE_parallel(result_m1a, thread = 16)
ode.m1b = qdODE_parallel(result_m1b, thread = 16)
net_m1a = lapply(ode.m1a$ode_result, network_conversion)
net_m1b = lapply(ode.m1b$ode_result, network_conversion)
#set seed to make same random layout
layout(matrix(c(1,2),1,2,byrow=TRUE))
set.seed(1)
network_plot(net_m1a, title = "Module1 a")
set.seed(1)
network_plot(net_m1b, title = "Module1 b")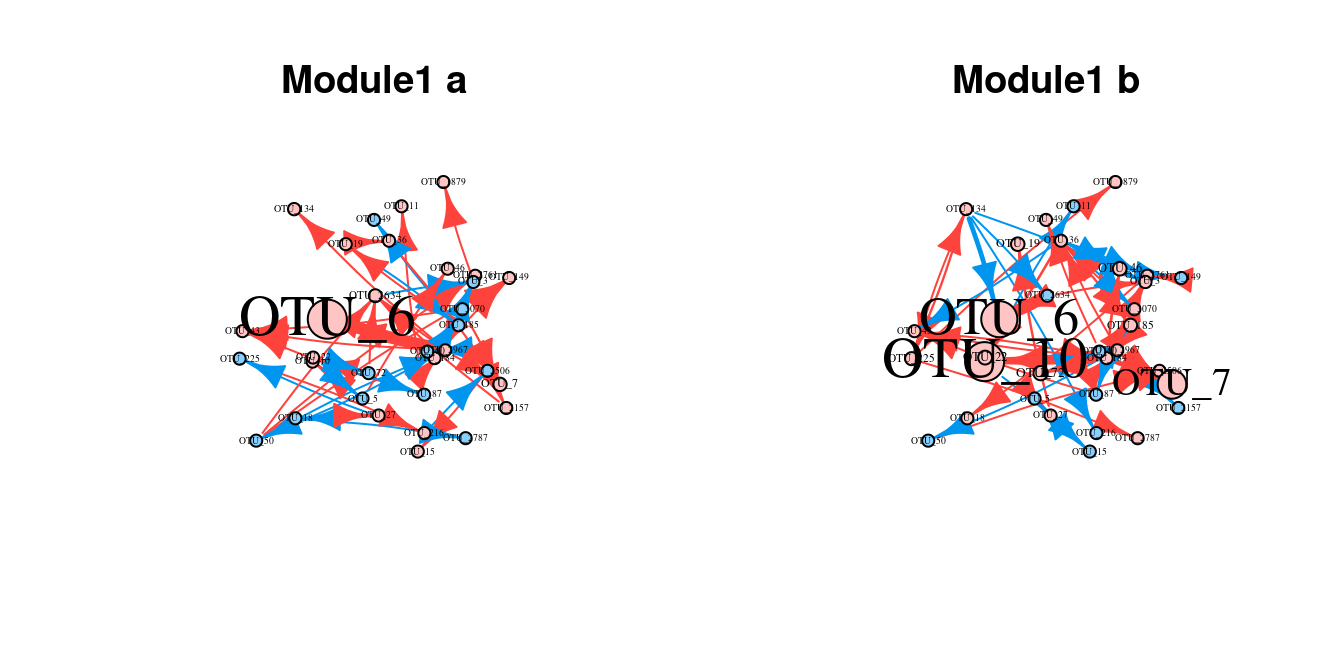
Troubleshooting
object “LL.next” not found
This happens when parameters optimization failure, try rerun
cluster.
plot failure when using fun_clu_plot() or
bifun_clu_plot()
This often happens when bad initial parameters is given and some cluster
is lost, try rerun cluster or use a smaller k.
Session info
sessionInfo()
#> R version 4.3.0 (2023-04-21)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 22.04.2 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] idopNetwork_0.1.2
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.4.6 utf8_1.2.3 shape_1.4.6
#> [4] stringi_1.7.12 lattice_0.21-8 digest_0.6.31
#> [7] magrittr_2.0.3 evaluate_0.21 grid_4.3.0
#> [10] iterators_1.0.14 mvtnorm_1.2-1 fastmap_1.1.1
#> [13] plyr_1.8.8 foreach_1.5.2 rprojroot_2.0.3
#> [16] jsonlite_1.8.5 glmnet_4.1-7 Matrix_1.5-4
#> [19] deSolve_1.35 survival_3.5-5 purrr_1.0.1
#> [22] fansi_1.0.4 scales_1.2.1 codetools_0.2-19
#> [25] textshaping_0.3.6 jquerylib_0.1.4 cli_3.6.1
#> [28] rlang_1.1.1 munsell_0.5.0 splines_4.3.0
#> [31] withr_2.5.0 cachem_1.0.8 yaml_2.3.7
#> [34] tools_4.3.0 parallel_4.3.0 reshape2_1.4.4
#> [37] polynom_1.4-1 memoise_2.0.1 colorspace_2.1-0
#> [40] ggplot2_3.4.2 orthopolynom_1.0-6.1 vctrs_0.6.2
#> [43] R6_2.5.1 lifecycle_1.0.3 stringr_1.5.0
#> [46] fs_1.6.2 ragg_1.2.5 pkgconfig_2.0.3
#> [49] desc_1.4.2 pkgdown_2.0.7 pillar_1.9.0
#> [52] bslib_0.4.2 gtable_0.3.3 glue_1.6.2
#> [55] Rcpp_1.0.10 systemfonts_1.0.4 highr_0.10
#> [58] xfun_0.39 tibble_3.2.1 knitr_1.43
#> [61] farver_2.1.1 patchwork_1.1.2 htmltools_0.5.5
#> [64] igraph_1.4.3 labeling_0.4.2 rmarkdown_2.22
#> [67] compiler_4.3.0